




우리나라「약사법」에서는 신약을 ‘국내에서 이미 허가된 의약품과는 화학 구조나 본질 조성이 전혀 새로운 신물질의약품 또는 신물질을 유효성분으로 함유한 복합제제의약품으로서 식품의약품안전처장이 지정하는 의약품’으로 정의하고 있습니다.
신약 출시를 위해서는 각국의 의약품 허가 당국이 규정한 기준을 충족하는 약의 안전성과 유효성을 증명하기 위한 많은 시간과 노력이 수반되는데요. 신약 개발 과정에 관한 기사를 보면 종종 이런 뉴스들을 자주 접하셨을 겁니다.
“A제약회사가 식품의약품안전처로부터 신약 후보물질의 IND(임상시험 계획 승인신청)를 승인받았다.”
“B제약회사는 9년간 개발해 온 신약의 임상 3상 결과를 바탕으로 美 FDA에 NDA(신약 허가신청)를 제출했다.”
뉴스에서 자주 언급되는 IND와 NDA. 용어는 들어본 적 있지만, 정확한 의미와 과정은 어렵게 느껴졌던 분들이 많으실 텐데요. 오늘 뉴스룸에서는 신약 개발의 필수 절차인 IND와 NDA에 대한 여러분의 궁금증을 풀어드리겠습니다!
■ “개발부터 시판까지” 신약의 개발 과정
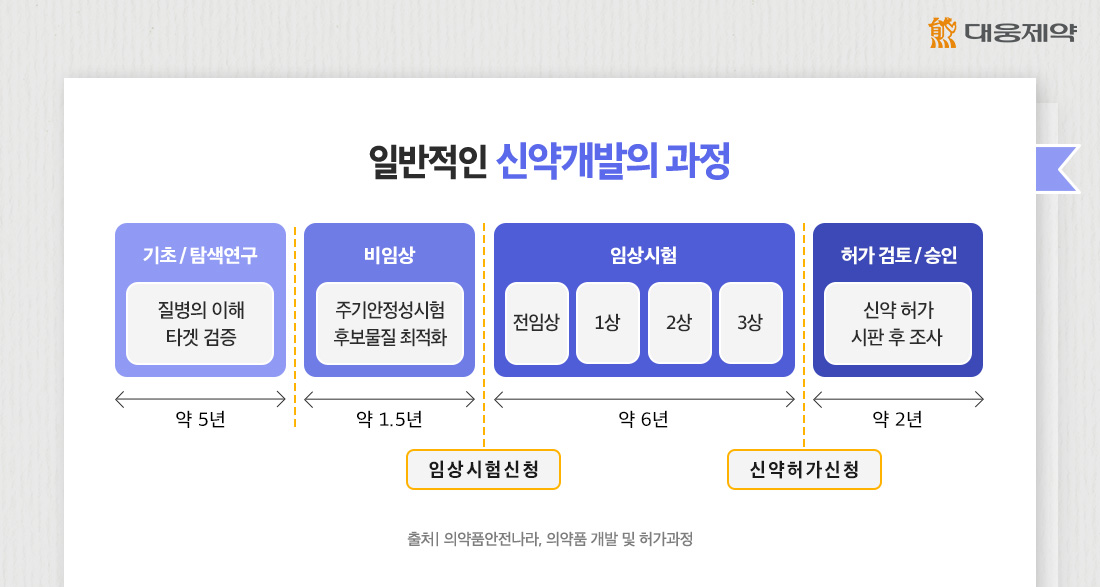
IND와 NDA에 대해 자세히 살펴보기에 앞서, 신약 개발 과정에 대해 간략히 살펴볼까요?
신약 개발 과정은 크게 발견 단계와 개발 단계로 나눌 수 있습니다. 일반적으로 발견 단계에서는 질병 타겟을 정하고 신약의 후보물질을 도출합니다. 개발 단계에서는 사람에서의 안전성과 유효성을 확인하는 임상시험을 실시합니다.
임상시험은 사람을 대상으로 하는 만큼 안전성과 윤리성이 보장되어야 하는 것이 가장 중요하겠죠. 때문에 제약회사가 임상시험을 실시하기 전에는 반드시 허가 당국으로부터 임상시험계획(IND)에 대한 승인을 받아야 합니다. 이후 1상, 2상, 3상 임상시험이 완료되면 이를 토대로 다시 허가 당국에 신약승인신청(NDA)을 제출해 승인을 받은 뒤 시판할 수 있습니다.
👉 관련 콘텐츠 보러가기
■ IND(임상시험 계획 승인신청)
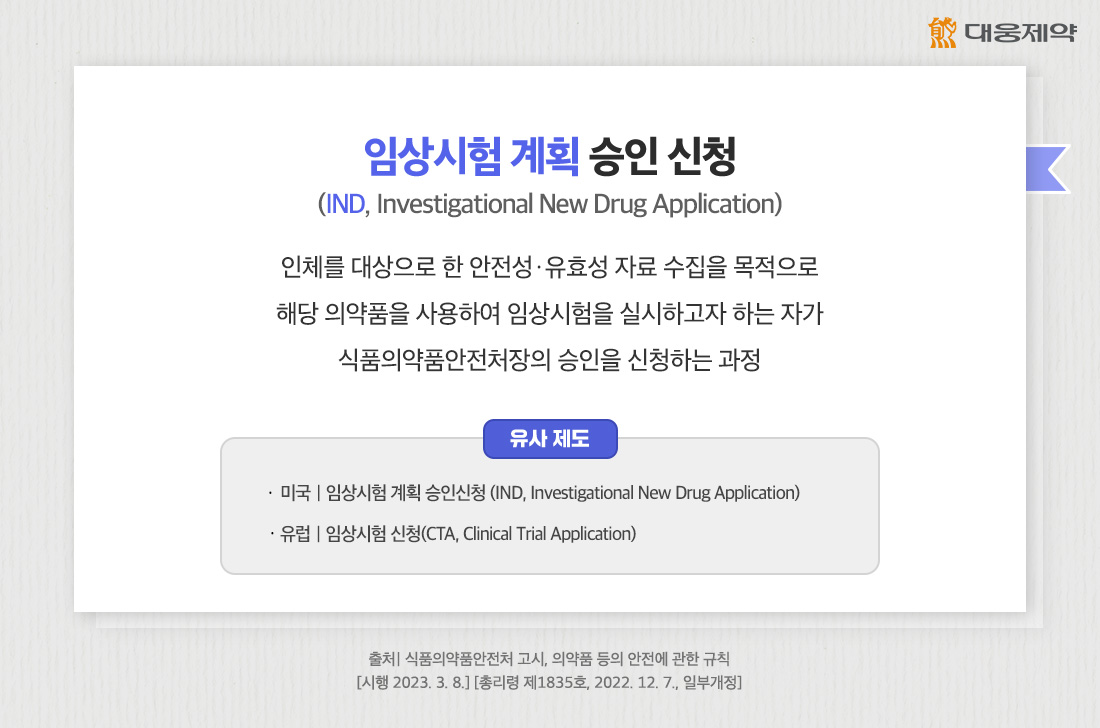
앞서 살펴본 것처럼, IND(임상시험 계획 승인신청)은 사람을 대상으로 임상시험할 수 있도록 허가 당국으로부터 공식적인 심사와 승인을 받는 절차를 말합니다.
우리나라 식품의약품안전처는 「의약품 임상시험 계획 승인에 관한 규정」을 통해 IND를 ‘인체를 대상으로 한 안전성ㆍ유효성 자료 수집을 목적으로 해당 의약품을 사용하여 임상시험을 실시하고자 하는 자가 식품의약품안전처장의 승인을 신청하는 과정’으로 정의하고 있는데요.
이와 유사한 제도로 미국은 FDA(미국 식품의약국)가 승인하는 IND(Investigational New Drug), 유럽은 EMA(유럽 의약품감독국)이 승인하는 CTA(Clinical Trial Application)가 있습니다.
□ IND(임상시험 계획 승인신청) 절차는 어떻게 되나요?
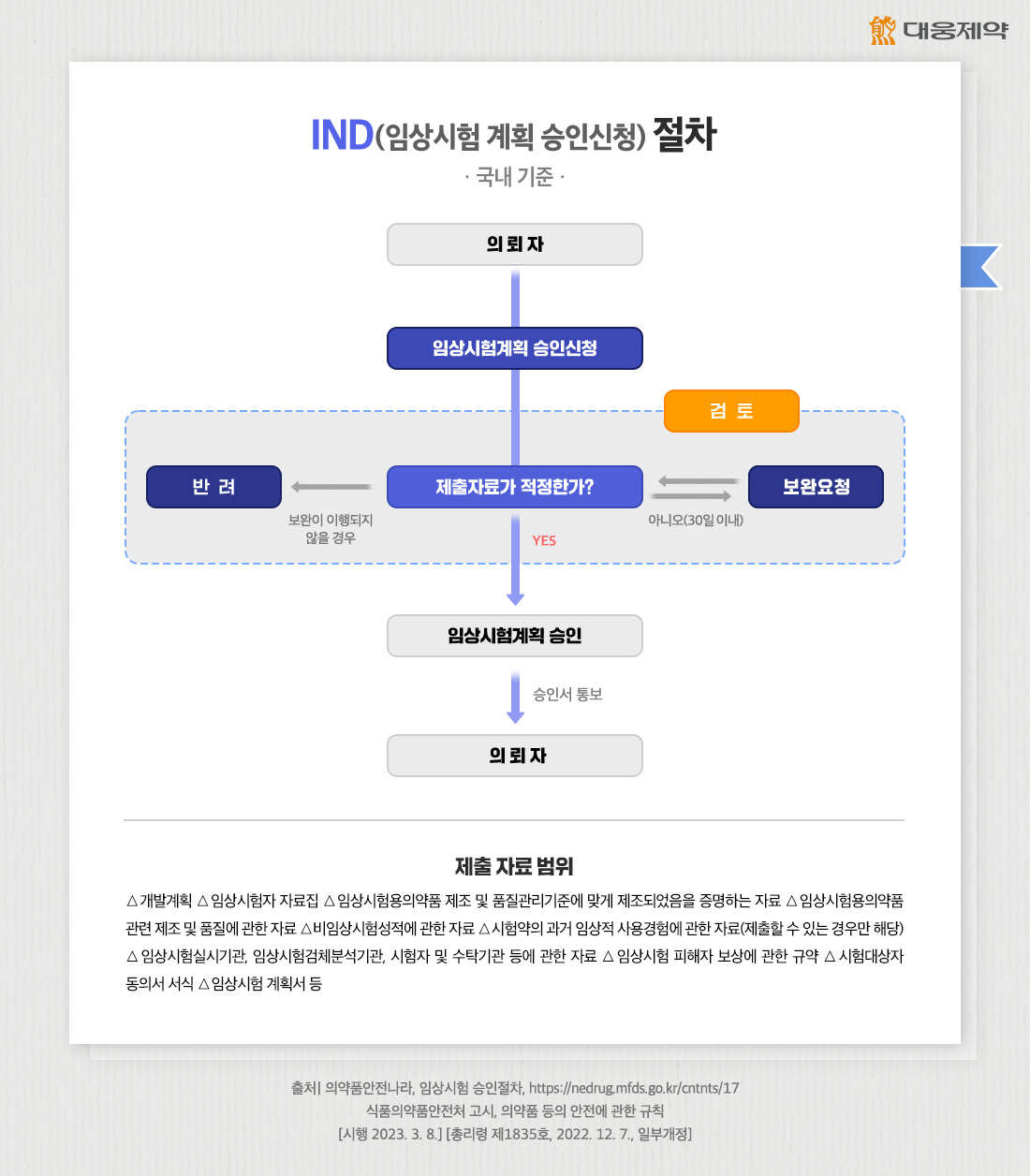
IND(임상시험 계획 승인신청)을 위해서는 허가 당국이 요구하는 자료를 제출해야 합니다. 우리나라 식품의약품안전처는 다음과 같은 자료를 제출하도록 요구하고 있습니다.
△개발계획 △임상시험자 자료집 △임상시험용의약품 제조 및 품질관리기준에 맞게 제조되었음을 증명하는 자료 △임상시험용의약품 관련 제조 및 품질에 관한 자료 △비임상시험성적에 관한 자료 △시험약의 과거 임상적 사용경험에 관한 자료(제출할 수 있는 경우만 해당) △임상시험실시기관, 임상시험검체분석기관, 시험자 및 수탁기관 등에 관한 자료 △임상시험 피해자 보상에 관한 규약 △시험대상자 동의서 서식 △임상시험 계획서 등
해당 자료를 토대로 안전성 및 유효성 평가가 완료되면, 의뢰자는 30일 이내 IND 승인 여부를 확인할 수 있습니다.
■ NDA(품목허가 승인신청)

NDA(품목허가 승인신청)는 임상시험을 마치고 의약품을 제조 및 출시할 수 있도록 허가 당국으로부터 공식적으로 승인 받는 절차를 말합니다.
우리나라 「약사법」에서는 ‘의약품을 판매하려는 경우에는 총리령으로 정하는 바에 따라 품목별로 식품의약품안전처장의 제조판매품목허가를 받거나 제조판매품목 신고를 하여야 한다’고 규정하고 있습니다. 따라서 신약을 포함해 국내 출시되는 모든 의약품은 식품의약품안전처의 승인을 받아야 합니다.
이와 유사한 제도로 미국은 FDA(미국식품의약국)가 승인하는 신약 허가신청(NDA, New Drug Application), 제네릭의약품 허가신청(ANDA: Abbreviated New Drug Application)이 있습니다. 유럽은 EMA(유럽의약품감독국)이 승인하는 판매 허가 신청(MAA, Marketing Authorisation Application)이 있습니다.
□ NDA(품목허가 승인신청) 절차는 어떻게 되나요?
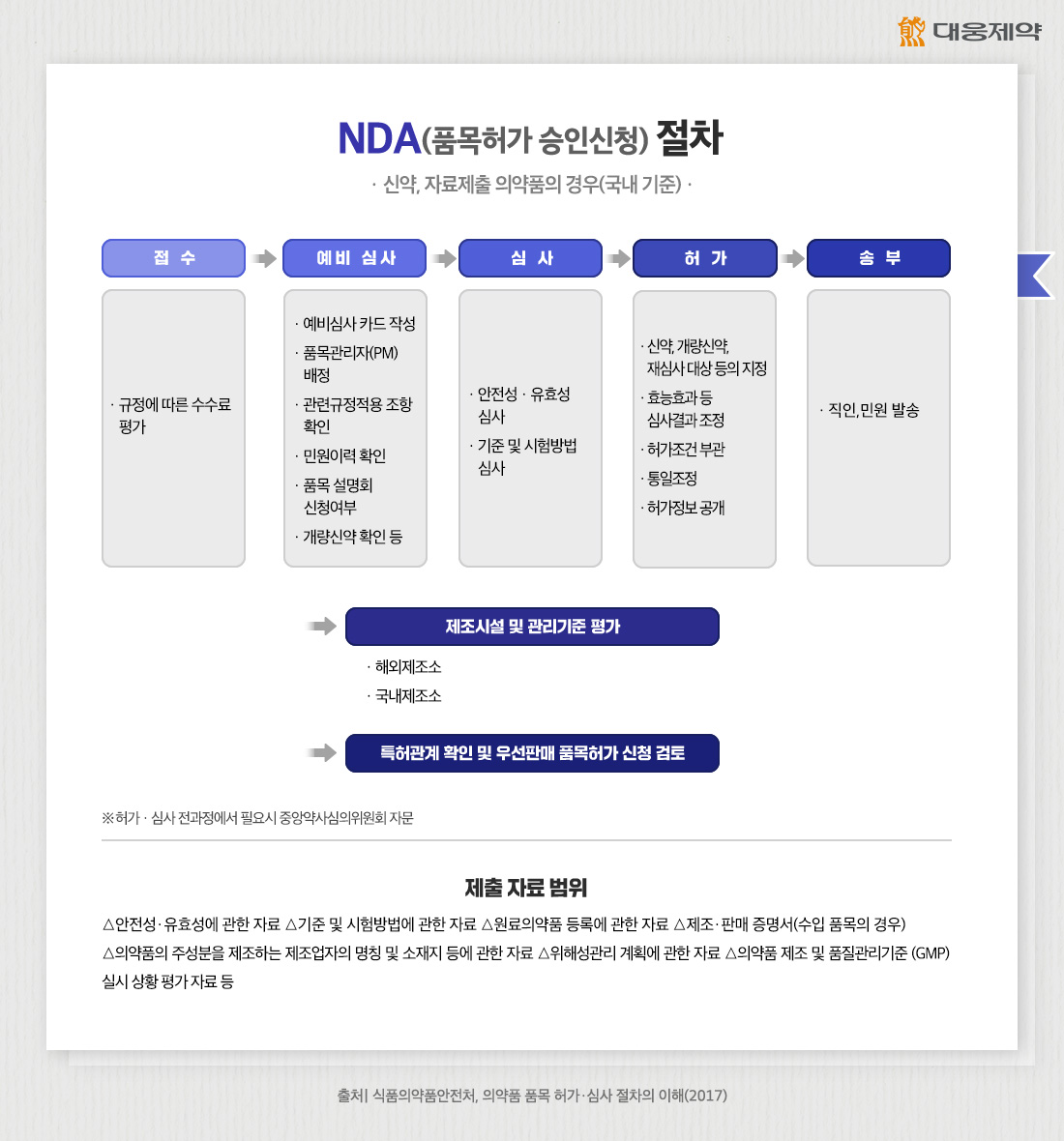
신약의 품목허가 신청 시에는 IND(임상시험 계획 승인신청)와 마찬가지로, 분야별 전문가들에 의해 절차가 진행됩니다. 예를 들면, 의료 담당자와 통계학자는 임상시험 데이터를 심사하여 임상시험의 방법과 신약의 유효성 및 안전성을 평가합니다. 약리학자는 동물을 대상으로 한 전임상 데이터를 심사하는 식으로 진행됩니다.
NDA(품목허가 승인신청)을 위해서는 허가 당국이 요구하는 자료를 제출해야 합니다.
신약의 경우 △안전성·유효성에 관한 자료 △기준 및 시험방법에 관한 자료 △원료의약품 등록에 관한 자료 △제조·판매 증명서(수입 품목의 경우) △의약품의 주성분을 제조하는 제조업자의 명칭 및 소재지 등에 관한 자료 △위해성관리 계획에 관한 자료 △의약품 제조 및 품질관리기준(GMP) 실시 상황 평가 자료 등의 제출이 필요합니다.
오늘은 그동안 생소하게 느껴졌던 임상시험 과정 속의 IND(임상시험 계획 승인신청)와 NDA(신약 허가신청)의 개념에 대해 알아보았습니다. 여러분들의 궁금증, 시원하게 풀리셨나요?
어렵고 딱딱하게 느껴지는 헬스케어 산업 용어를 보다 쉽고 재미있게 이해하고 싶다면, 앞으로도 자주 대웅제약 뉴스룸에 방문해 주세요. 🙂
[관련 콘텐츠]

